
Nytasha was worried when the nurses whisked away her daughter moments after she was born. They said the baby was anemic and had jaundice. There could be something wrong with her liver.
But a blood test soon revealed Nytasha’s daughter, Za’Mya, was born with sickle cell anemia, one of the most common inherited blood disorders in the United States. It can occur in all races but is most common in African Americans. One out of 365 African American babies born in the U.S. has sickle cell disease.
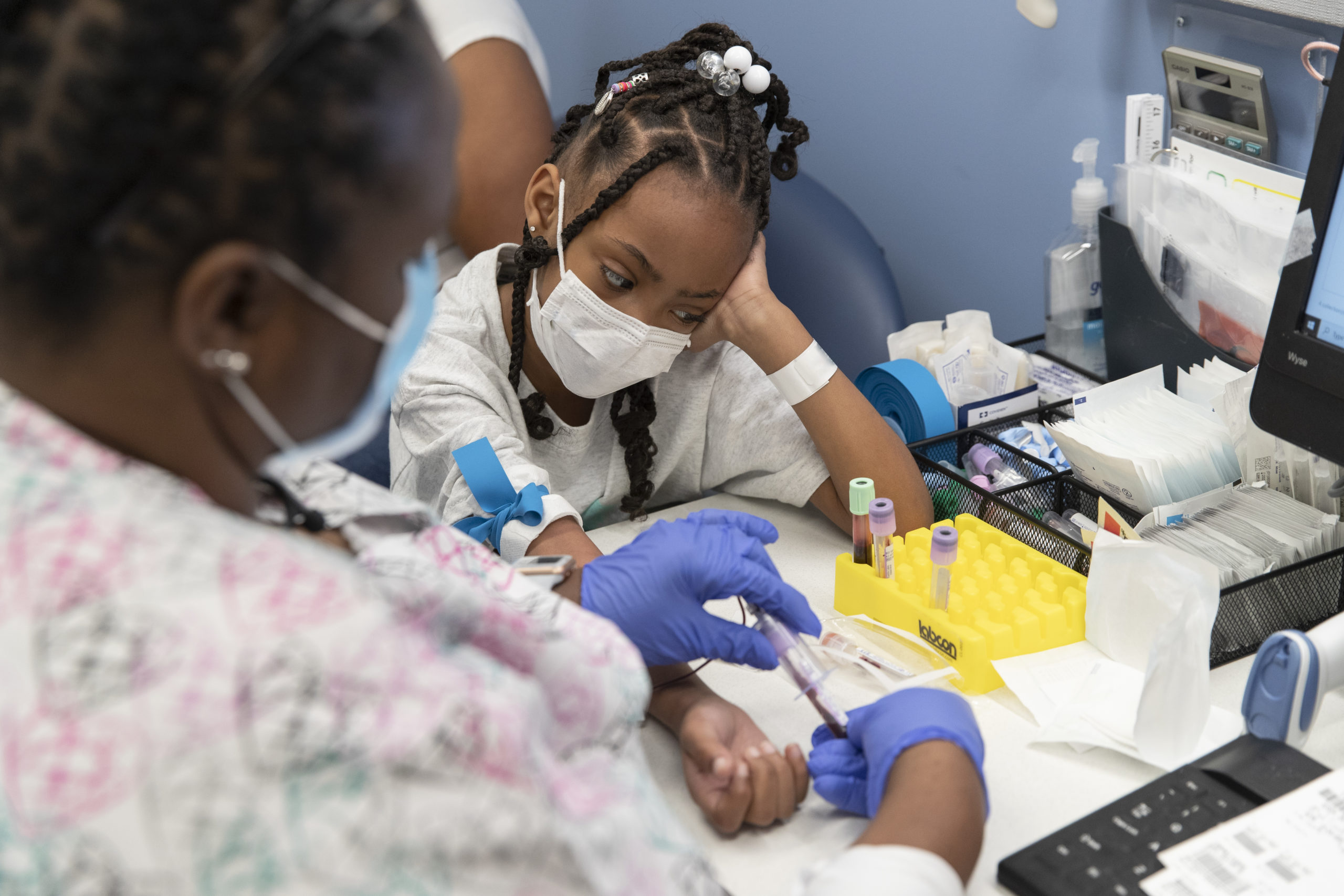
Za’Mya, who is now 7, was diagnosed in the first week of her life and was referred for care and treatment at St. Jude Children’s Research Hospital, which runs one of the largest sickle cell programs in the country, treating about 900 children a year.
Nytasha was aware she carried the sickle cell gene, but knew little about the disease, which forms hard fibers inside red blood cells causing them to distort from healthy, round and plump into sickle or banana shapes. The abnormally shaped red blood cells cannot move through blood vessels easily, so they get stuck, preventing the crucial delivery of oxygen to some organs and tissues and causing debilitating, sometimes life threatening, pain crises.
For Za’Mya, the pain comes unexpectedly and throbs along her legs, but sometimes in her chest, too, forcing the family to rush to the hospital for urgent treatment.
Over the last 60 years, doctors and researchers at St. Jude have set the stage to dramatically improve the care and understanding of sickle cell disease.
It was on the campus in Memphis where an African American doctor, Rudolph Jackson, developed community outreach programs and clinical protocols serving sickle cell-affected families in 1968. In a few years, Jackson’s pioneering work had amassed such recognition and stature that the federal government asked him to help establish a national protocol for the treatment and care of sickle cell patients.
A St. Jude patient was the first in the world to be cured of sickle cell anemia through a stem cell/bone marrow transplant. And work led by scientists and researchers at St. Jude also showed that using the drug hydroxyurea daily could boost average hemoglobin levels, thus severely reducing the frequency of hospitalization for pain crises.
Recently, St. Jude partnered with other top institutions in the country to develop and refine novel gene therapies that would allow doctors to edit the genes responsible for sickle cell in fetuses, allowing babies prone to the disease to be born healthy. Published early results of gene editing studies are “very promising,” said Dr. Clifford Takemoto, Director of Clinical Hematology at St. Jude and Lemuel Diggs Endowed Chair in Sickle Cell Disease.
Takemoto added that other screening measures have also been developed that identify children at risk for other major life-threatening conditions such as stroke. So now, therapies can be implemented before children experience such devastating complications. Using these new therapies and screenings, doctors and scientists have markedly reduced the chances of early death and disease caused by sickle cell.
Still, there is much work left to do to improve the lives of the more than 100,000 patients affected by sickle cell in the U.S., and the tens of thousands with it around the world, particularly in Africa, Takemoto said.
“This is a condition where the life expectancy is still in the mid40s,” he said.
These realities weigh on Nytasha’s mind. She asks lots of questions at clinic visits about Za’Mya’s prognosis, which looks encouraging now. Nytasha has big dreams for her daughter.

“She’s going to get married. She’s going to have me some grandbabies. We are going to take trips. She’s going to be a doctor. She’s going to do everything she wants to do…and more,” Nytasha said.

{kind=link}